Thalidomide’s acceptance in Europe and rejection in the U.S.
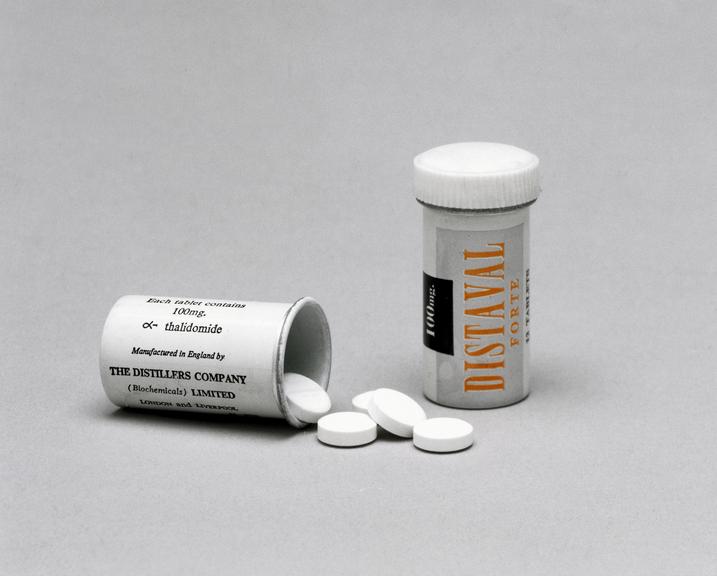
In the 1950s, a West German pharmaceutical company brought the drug thalidomide to the European market as a sedative. Thalidomide quickly gained approval in over 40 countries under a multitude of brand names – treating everything from sleeplessness to the common cold, as well as morning sickness during pregnancy. Despite thalidomide’s success abroad, the Cincinnati-based pharmaceutical company Merrell had a difficult time introducing it to the U.S. drug market.
Dr. Frances Oldham Kelsey was recently hired as a medical reviewer at the FDA when she was assigned to thalidomide’s new drug application and tasked with assessing the clinical trial data. In an autobiographical document held by the FDA, Dr. Kelsey noted that thalidomide’s application wasn’t delayed because it was found to be unsafe, rather that Merrell had done very little to prove that it was safe. The clinical trial section mostly contained physician testimonials, glowing claims that weren’t supported by trial data, and a distinct lack of data concerning nerve-damage side effects observed in European patients.

At the time of thalidomide’s FDA review process, it was considered an astonishingly safe drug. Dr. Kelsey and the FDA were pressured to quickly approve the drug, but they refused until Merrell could provide adequate data on its side effects – particularly during pregnancy. At the same time, rising numbers of birth defects were observed abroad with no known cause. Eventually, an Australian obstetrician, Dr. William McBride, made the link between babies delivered with severe limb deformities and his prescriptions for thalidomide. In 1961, Dr. McBride published a letter describing his observations in the British journal, The Lancet, causing hospitals to pull thalidomide from their shelves and leading Merrell to formally withdraw their application from the FDA.
Despite its lack of approval, patients in the United States could get thalidomide while traveling abroad or by participating in the U.S.-based clinical trials. However, the FDA’s refusal to approve the drug limited the damage, and only eighteen cases of birth defects were definitively linked to thalidomide in the U.S. As a result of the scandal, Congress passed the 1962 Kefauver-Harris Drug Amendments that created strict requirements for pre-clinical trial evidence and study design, regulated how companies could market their drugs, and established the concept of informed consent in clinical trials. Dr. Kelsey earned the President’s Award for Distinguished Federal Civilian Service and had a prominent position in the newly restructured FDA.
Uncovering thalidomide’s target and repurposing it for a new class of drugs
Despite its notoriety, thalidomide is still used for treating multiple myeloma and leprosy under very strict control. However, the mystery of its role in causing birth defects remained murky until 2010, when a study identified the protein Cereblon as a direct target of thalidomide. Researchers found that Cereblon works as an E3 ligase – meaning it tags other proteins to mark them for destruction – and that thalidomide disrupts that activity, causing deformities in zebrafish and chicks. A later study discovered that thalidomide forces Cereblon to tag a critical protein called SALL4 that turns on the genes responsible for limb development, providing a molecular reasoning behind the thalidomide scandal of the 1960s.
E3 ligases have been the center of attention in a rapidly developing area of therapeutics known as targeted protein degradation, which involves hijacking E3 ligases to mark disease-causing protein “targets” for destruction. A popular way to accomplish such a feat is through a PROTAC, or proteolysis-targeting chimera. In PROTACs, one half binds the E3 ligase and the other half binds the target, bridging the gap between the two proteins and giving the E3 ligase an opportunity to destroy a protein that causes disease. Traditional drugs often work by binding the small, specific pocket that gives a protein its activity, thereby shutting it down. However, not all troublesome proteins have such pockets and are considered “undruggable” by conventional drug design. PROTACs can avoid this issue by binding anywhere on the target, blowing the field wide open to target notorious proteins long-thought to be “undruggable”.
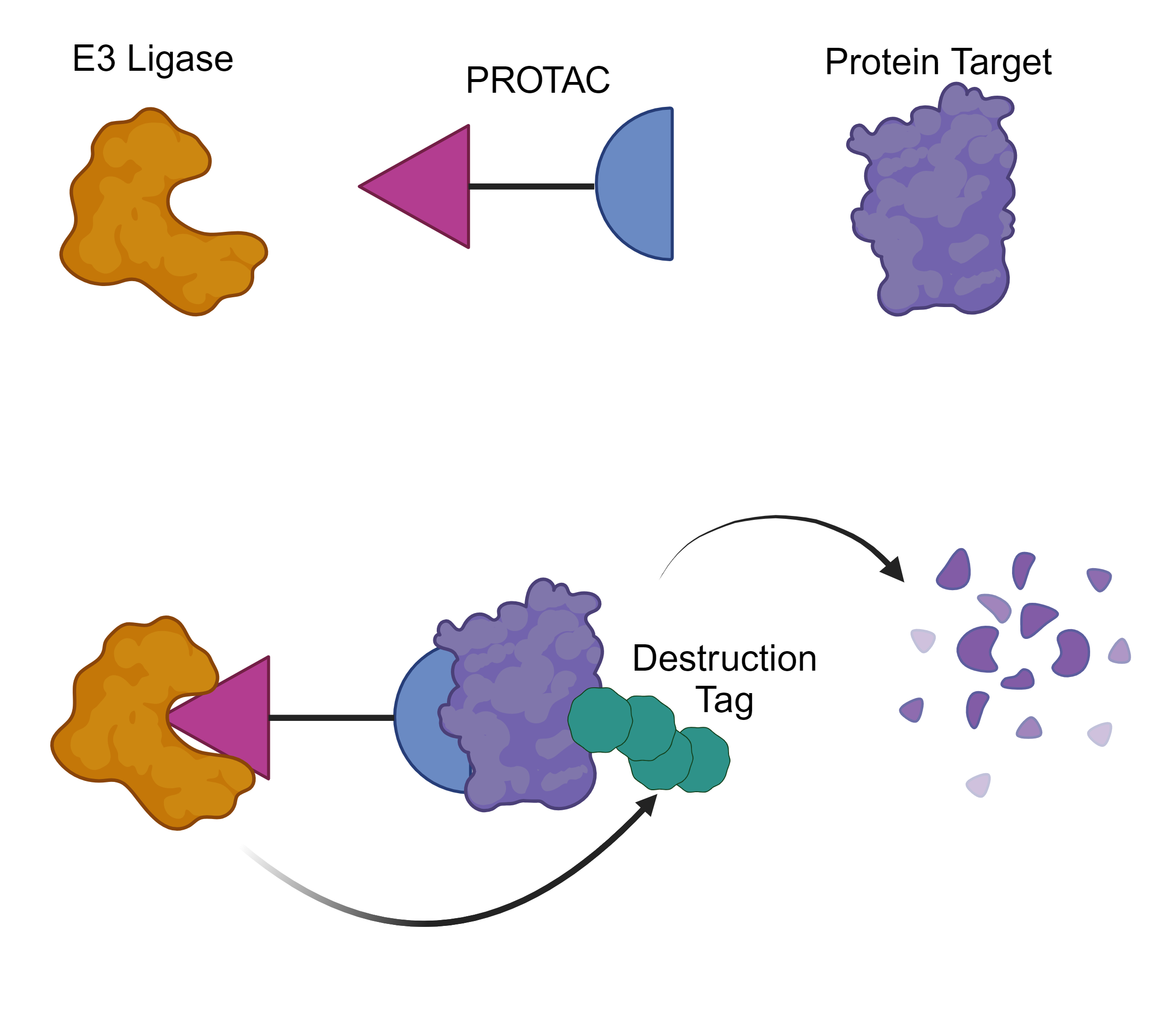
The concept of a PROTAC was first presented in 2001. Over a decade later, the first set of PROTAC designs targeting well-known, disease-causing proteins were unveiled. Using its interaction with thalidomide, Cereblon remains one of the major E3 ligases harnessed for PROTACs today. As of January 2023, at least eighteen PROTACs have entered clinical trials. Two such PROTACs – both of which hijack Cereblon – are in Phase II or III clinical trials and take their aim at well-established protein targets in prostate and breast cancer.
Major concerns for PROTACs include their structure and size, which are unlike traditional drugs, as well as their potential to destroy the target protein in healthy tissues. A recent study investigated the opposite side of the PROTAC. Researchers scoured the hundreds of E3 ligases to identify 76 that could be successful in new PROTAC designs based on whether they have known binder molecules, are less abundant in healthy tissue, are highly abundant in cancerous tissue, and more. By having an expanded repertoire of E3 ligases at their disposal, scientists can design PROTACs tailored to a disease and minimize the chance of side effects. Even as the targeted protein degradation field turns its gaze towards other E3 ligases, PROTACs that harness the unique interaction between Cereblon and thalidomide continue to pave the way through clinical trials as the leaders in this new class of therapeutics.
Peer Editors: Caitlyn Molloy & Savannah Muron