Structure equals function. It’s one of the foundational principles of biology. Whether it’s making muscles contract, transmitting information between neurons, or converting sunlight and sugar into energy, the function of a protein is determined by its three-dimensional shape.
Knowing protein structure is enormously useful if you want to design a drug that will help a malfunctioning protein, or re-engineer existing proteins to perform novel functions like produce biofuels or degrade plastics. Biologists have long dreamed of obtaining a deep enough understanding of protein structure to make these types of applications commonplace. While a great deal of progress has been made, the bottleneck has always been that experimentally solving a protein’s structure is slow and expensive work.
In theory though, a protein’s structure could be computationally predicted, or at least guessed at within an acceptable margin of error, using only its unique sequence of amino acids. But for the last half century, making accurate structural predictions of proteins has remained one of the great puzzles of modern biology. While there have been major advances, particularly when it comes to predicting small parts that are commonly found among certain protein families, the goal of reliably modeling whole protein structures has remained out of reach.
Now, a new artificial intelligence method called AlphaFold, developed by a sister company of Google called the DeepMind group,has made a significant leap forward in predicting protein structure. Recently published in the journal Nature, AlphaFold improves on existing approaches by using machine learning methodologies adapted from applications in computer vision and language interpretation. The new method obtained a median prediction accuracy of more than 90% in the 2020 Critical Assessment of protein Structure Prediction (CASP) competition, considered the gold standard for testing new modeling techniques. As a comparison, the next best method from previous CASP competitions was an earlier version of AlphaFold that only got a median accuracy of ~60%.
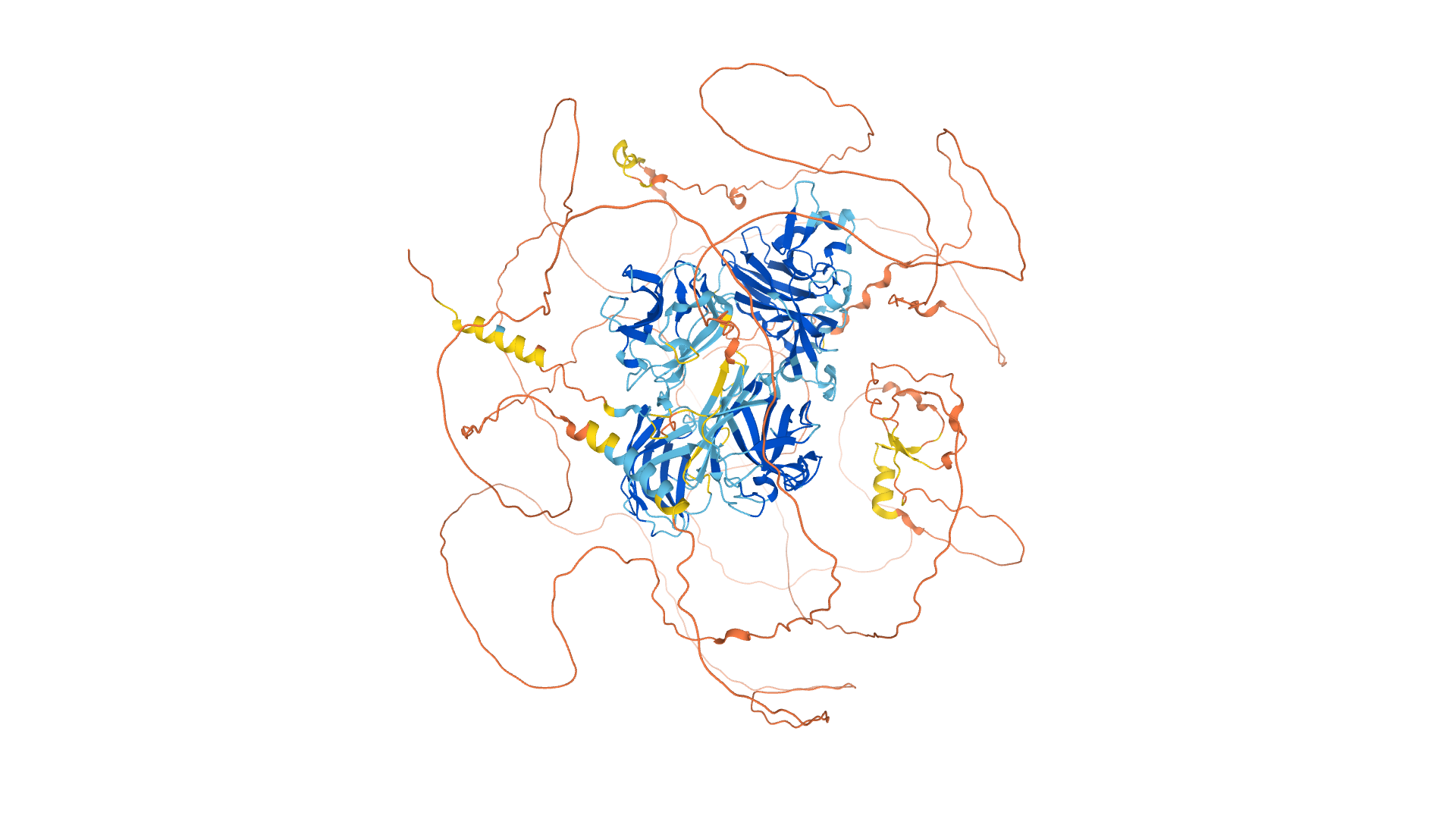
AlphaFold is built with two algorithms that are able to communicate and iteratively refine the model. One algorithm mines existing databases to find similar amino acid sequences in different organisms and builds a graph of the similarities. Making these types of alignments can be informative when modeling structure because they highlight what parts of a protein are the least variable over evolutionary time and thus most likely to be functionally important.
The other algorithm uses these alignments to make predictions about how pairs of amino acids relate to one another in space, generating a predicted structure. This predicted structure is then recycled back into the network to further refine the alignments and the final model.
Last week the AlphaFold team, in partnership with the European Molecular Biology Laboratory’s Bioinformatics Institute (EMBL-EBI), announced the launch of a public database of more than 350,000 predicted protein structures including a majority of all human proteins and those of many model organisms commonly used in research. Writing on the company blog on the day of the announcement, DeepMind CEO Demis Hassabis stated that “our dream is that AlphaFold, by providing this foundational understanding, will aid countless more scientists in their work and open up completely new avenues of scientific discovery.” While the ultimate impact of the AlphaFold software and public database are yet to be seen, it is a clear demonstration of how machine learning and artificial intelligence can be used to address seemingly insurmountable scientific challenges .
Peer Editor: Rami Major