T cell-based therapies, or “living drugs” as coined by Dr. Carl June, utilize the potent killing activity of T cells, an arm of the immune system, to target cancers. In the early stages of T cell-based therapy, T cells were isolated from tumors, expanded ex vivo, selected for specific anti-tumor clones, and infused back into the patient. Nowadays, T cell products are genetically modified to express receptors to more specifically target cancers with better persistence in patients. So how are these “living drugs” manufactured?
Here at UNC, a Good Manufacturing Practices (GMP) facility housed off of NC-54 generates all the T cell products used in phase I/phase II clinical trials by the Lineberger Comprehensive Cancer Center. These facilities are regulated by the US Food and Drug Administration under the authority of the Federal Food, Drug, and Cosmetic Act.

I spoke with Paul Eldridge, PhD, Director of the UNC Lineberger Advanced Cellular Therapy Facility, to learn more about how GMP facilities work. Dr. Eldridge was recruited in 2014 by the Lineberger Comprehensive Cancer Center, which was interested in starting a cellular immunotherapy program and building a GMP facility. Dr. Eldridge’s personal interests are in chimeric antigen receptor T cells (CAR-Ts) and hematopoietic stem cells, with a focus in cancer immunotherapy.
An excerpt of our conversation is below, edited for clarity:
Lee Hong (LH): What products are manufactured at UNC’s GMP facility?
Paul Eldridge (PE): Here at UNC, we focus on advanced research products. The FDA divides cell products into minimal manipulation and more than minimal manipulation. Minimal manipulation essentially does not change the character of the cell, which means you can isolate, purify, or freeze the cells. More than minimal manipulation involves putting cells into tissue culture.
LH: Huh, why is that?

PE: Well, when you put cells into culture, they are dividing, experiencing a different stimulus in the culture medium, and may differentiate into other cell types. In other words, anything that could potentially change the innate nature of the cell is considered more than minimal manipulation. Certainly gene manipulation would be included here as well. How you intend to use the cell products, what the FDA calls “homologous use,” also matters. If the investigator is intending to use the cells in a manner that it is not normally functioning (i.e. non-homologous use), the FDA kicks those products up to a higher regulatory environment and calls them more than minimal manipulation.
LH: So at UNC, are most of the advanced research products you work on derived from peripheral blood?
PE: Yes, we mainly manufacture CAR-T cells from peripheral blood. We are also working with another investigator, Shawn Hingten, who is using skin fibroblasts. Outside of UNC, other investigators are using adipose-derived stem cells or mesenchymal stem cells common in regenerative medicine.
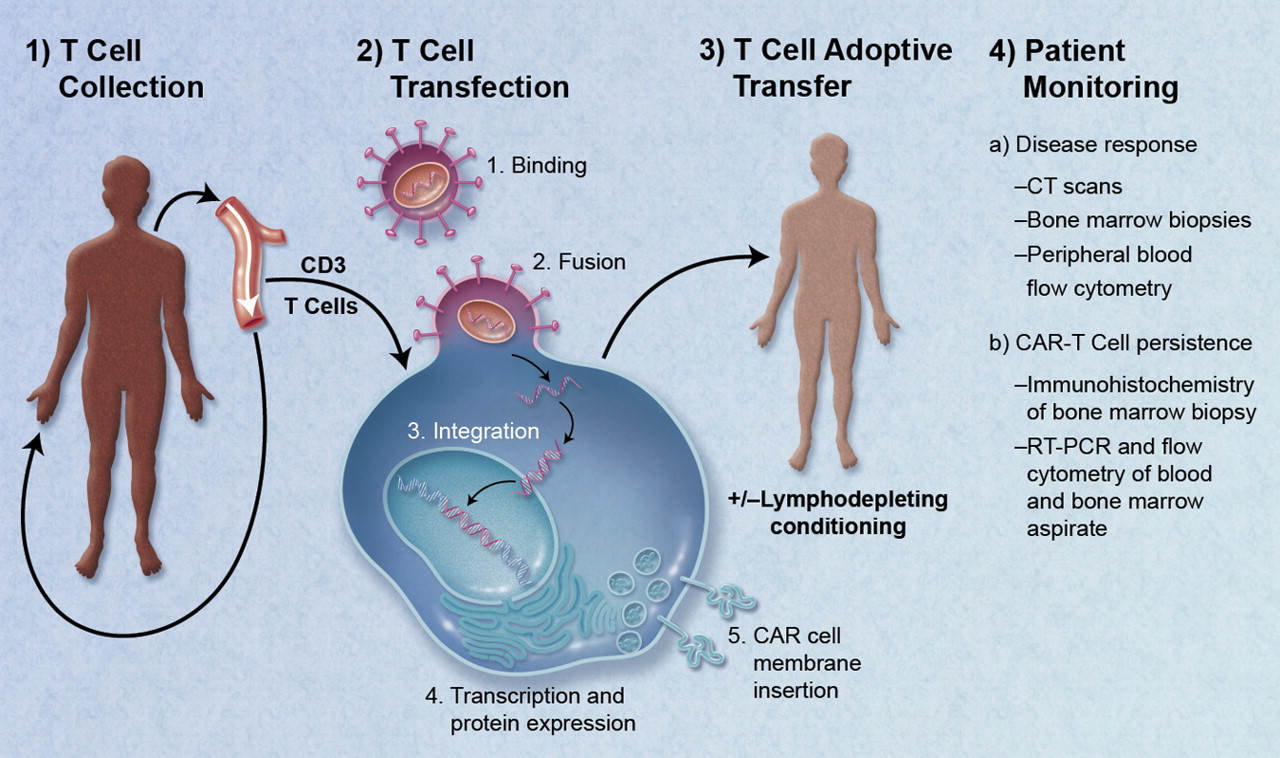
LH: How is UNC’s GMP facility set up?
PE: The facility is 5,000 square feet, with half of the space as clean room facilities. We have six separate processing rooms, five for patient samples and one with a different air system for virus protection. It’s a ISO-7 environment, meaning we use “bunny suits” and have to re-gown each time we enter or leave a room.
Patient rooms are positive air-pressured to the hallway in order to minimize anything coming back into the room. The air is 80% recirculated. In contrast, the virus room is negative pressure and the air is 100% single-pass filtered with no recirculation.
LH: Oh wow, there are a lot of details involved here.
PE: Yes, part of building a GMP facility is paying attention to construction details. We designed ours for an academic center, which is a different layout than that for pharmaceutical manufacturing.
LH: So how much of what you do in a GMP facility is automated versus manual?
PE: It really depends on where you are in product development. In our facility, we are focused on early phase I trials in which we have not nailed down a manufacturing process so we are pretty manual in most of our applications. Part of what we’re doing is learning how to manufacture the cells we need with minimal effort in a system that is as closed as possible.
As we move to phase II, then we start looking at scaling up due to the need for more cells. This is where bioreactors can be helpful and the steps become more automated. Cell therapy is where drug manufacturing was 75 years ago, in the sense that not much is automated. But nowadays, the technology is continually advancing. Miltenyi is offering a bioreactor called CliniMACS Prodigy that makes it sound as easy as pushing a button.

LH: What sort of training and skill sets are needed for someone to work at a GMP facility?
PE: It breaks down into a couple of areas. One is whatever the process requires, in this case usually tissue culture (TC). We do a lot of TC as part of manufacturing cells. Another area is in regulated, quality control testing. We do a lot of characterization analysis on our cell products. We establish release specifications for every product we make so we have to do all the assays before patients can receive them. These assays aren’t necessarily done in the GMP facility, just wherever we can do it most easily.
The important thing is that all the assays need to be performed in a more GMP manner than you might encounter in a basic research lab. Documentation, Standard Operating Procedures (SOPs)…we do everything by SOPs. This is because we need to trace all of our materials, use everything within its expiration date, and keep up with instruments for calibration and maintenance. We also train people on site on whatever they’re doing, document the training, and ensure trainees maintain competency for quality control testing. In other words, we do all the tests you see in a typical research lab but in a more stringent, reproducible, and regulated manner.
Another important skill is learning how to work in a clean environment. Everyone thinks they know how to use a biological safety cabinet (BSC), but there are good ways and bad ways, and there are ways you have to operate when you are trying to minimize the risk of cross contamination. So we do a lot of cleaning, and we have to document everything we do.
In general, I don’t necessarily need someone with PhD credentials but I do need someone who is smart, dedicated, and extremely detail-oriented. We are looking more for personality and attitude than specific qualifications.

LH: How are careers and/or skills used at GMP facilities in academic centers different than in pharmaceutical companies?
PE: Well, in an academic center you see everything, and that’s the most enjoyable part of it. We will often break out into more research and development (R&D) work as opposed to hands-on, clean manufacturing work. People float back and forth between what they are comfortable doing. I have people with PhDs and high school degrees working in the facility.
From the industry side of things, a very different set of skills is needed. That’s because by the time you get to phase II/III clinical trials, the process is set and there is no situation where you’ll be making changes. Of course, you still need to have attention to detail and be thorough, but the important aspect is to follow the instructions and nothing else. However, when something doesn’t work, you’ll need enough wherewithal to understand whether it was a process accident or a random occurrence.
LH: Finally, where do you see cell therapy going in the next 25 years?
PE: It’ll become more rote, with more big pharma involved. The current model, as long as we are talking about autologous starting material (i.e. cells from the same patient), is not really scaled up so much as scaled out. There are still individual batches (or lots) made for individual patients. Where will we do this? It’s still not clear where it is economically advantageous to do so.
For example, Novartis has a centralized manufacturing facility [for Kymirah]. That works fine for now, but will Novartis keep up with material demands? It’s not just tissue culture media, they have to make lentiviral vector, and the suppliers right now can turn out product for only 25-30 patients at a time. No one has ever tried making 10,000 personalized products before. Moreover, the FDA requires lentiviral vectors to have a shelf life of couple years so vector suppliers are desperately trying to scale up. We are still in the early wild west stage but it is fascinating.
All pictures provided by Dr. Eldridge.
Peer edited by Justine Grabiec and Erin Langdon.
Follow us on social media and never miss an article: