Huntington’s Disease
In 1872, Dr. George Huntington described a disease that he termed ‘chorea’ – a whimsical name that comes from the Greek for ‘dancing in unison’. Huntington chose the name “on account of the dancing propensities of those who are affected by it.” The disease, as he described it,
“is essentially a disease of the nervous system. Its most marked and characteristic feature is a clonic spasm affecting the voluntary muscles…the will is there, but its power to perform is deficient, the desired movements are after a manner performed, but there seems to exist some hidden power, something that is playing tricks, as it were.”
Today, we know chorea as Huntington’s disease, and there is nothing whimsical about it. Instead, it is a devastating illness in which the striatum, a part of the brain involved in movement, is slowly destroyed, leading to loss of motor function as well as cognitive decline. Most people inherit Huntington’s disease from a parent, meaning that a child watching their parent decline carries an additional burden – there is a 50% chance that they will suffer the same fate. And if they do, it is likely that their symptoms will start earlier and be more severe.
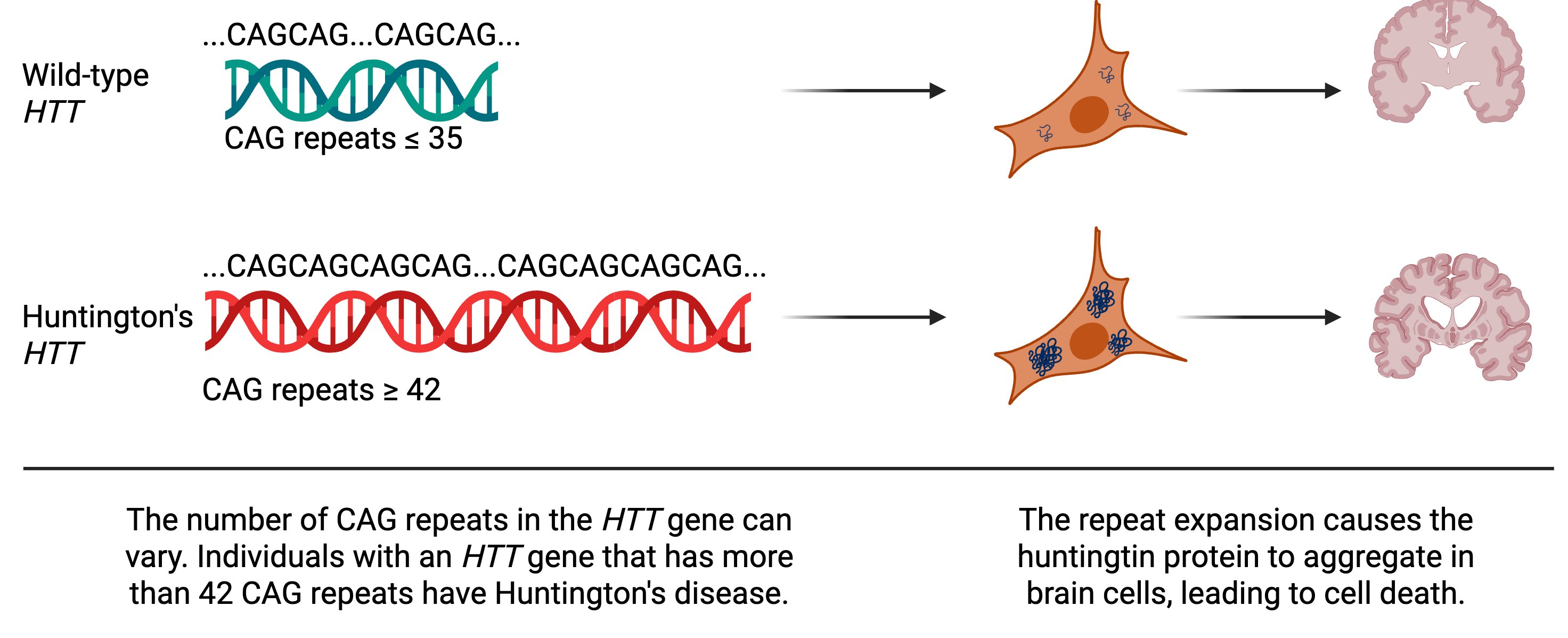
Huntington’s disease is caused by an altered form of a gene called huntingtin or HTT. Every gene is made up of a series of chemicals called nucleotides that are represented as A, C, T, or G. For HTT, the nucleotide sequence encodes the instructions for making a protein (also called huntingtin). In its normal or “wild-type” form, part of HTT’s sequence is naturally repetitive, containing up to 35 copies of the sequence CAG. Typically, DNA replication, the process in which cells copy DNA to make more cells, is amazingly accurate with cells only making one mistake for every one hundred million nucleotides. But repetitive regions, like those found in HTT, can serve as a molecular tongue twister, causing the cell to stumble and insert extra CAG repeats. And a person whose HTT gene has more than 42 CAG repeats is guaranteed to get Huntington’s disease.
Each CAG repeat in HTT encodes an amino acid (a building block for a protein) called glutamine. Glutamine is not inherently dangerous – it’s essential for our bodies to function – but it’s also “sticky”, and when a protein has too many glutamines in a row, the protein starts to stick to itself. In Huntington’s disease, the huntingtin protein sticks to other copies of itself and forms aggregates in brain cells, trapping other important proteins, and ultimately destroying the cell.
A Promising New Therapy
For over 150 years, since Dr. George Huntington first gave the disease a name, there has been no way to cure, slow, or prevent Huntington’s disease. At best, some of the symptoms can be lessened by taking medicines that help control movement. For years, the great white whale of Huntington’s disease research has been a therapy that can target the HTT gene directly to reduce the amount of toxic protein that is produced. And last month, uniQure, a gene-therapy company, released data from a clinical trial showing that a gene therapy can dramatically slow progression of Huntington’s disease.
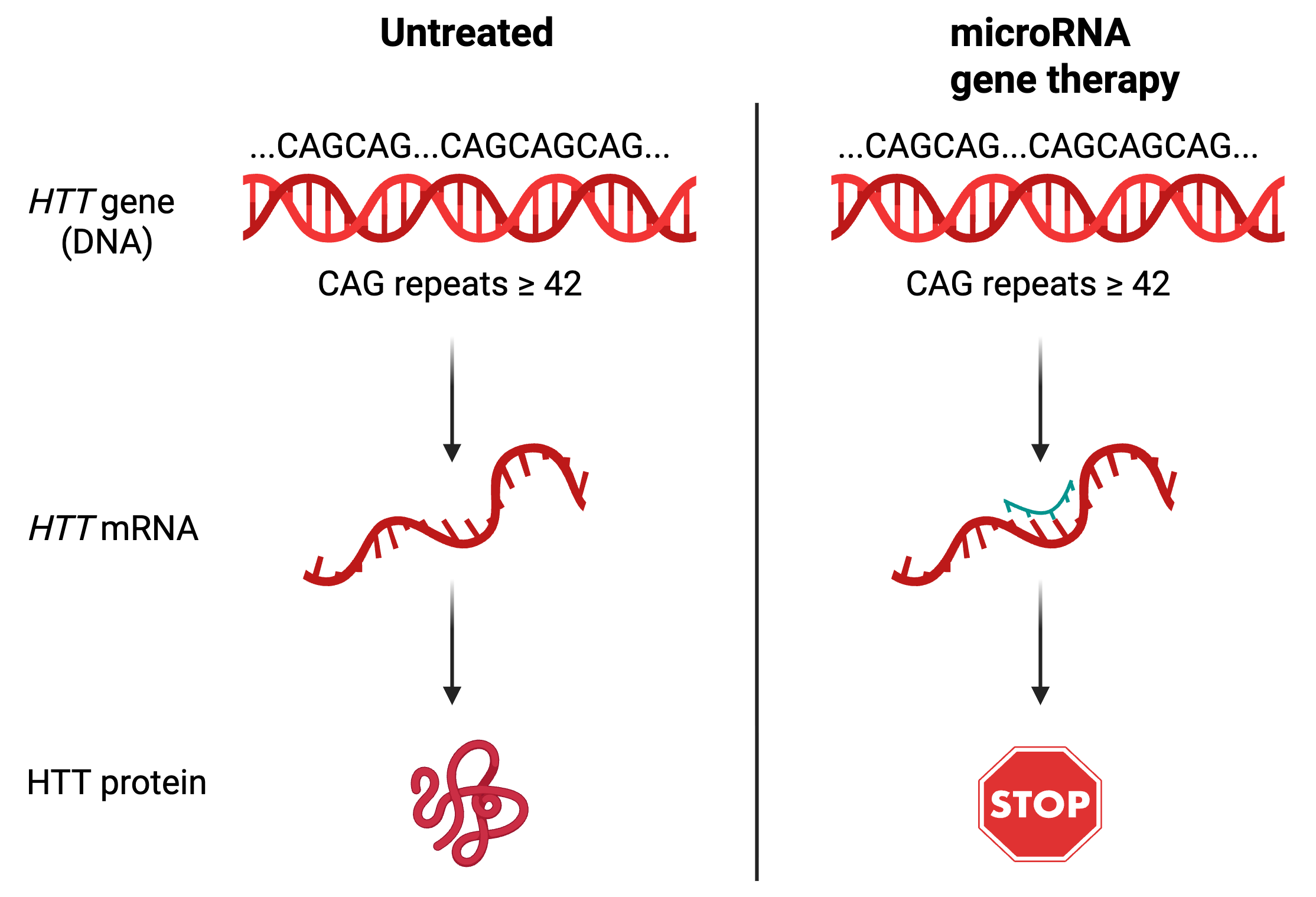
The gene therapy that uniQure designed targets the HTT messenger RNA (mRNA) – an intermediate between the expanded HTT gene and the toxic protein it produces. Usually, the sequence of a gene is directly copied into mRNA, which goes to another part of the cell to be turned into a protein. By targeting the HTT mRNA, the huntingtin protein will never get produced. To do this, uniQure turned to microRNAs, a mechanism for regulating mRNAs that won the Nobel prize in 2024. Briefly, microRNAs are very short RNA sequences, and when the nucleotide sequence of a microRNA matches the nucleotide sequence of a messenger RNA, it attaches to the mRNA and prevents protein synthesis. In this case, scientists at uniQure designed a microRNA that will specifically target the disease-causing version of HTT, preventing the production of the expanded huntingtin protein.
In uniQure’s clinical trial, 29 patients were given either a low dose or a high dose of the gene therapy. Over three years, 12 patients who received the high dose saw their disease progression slow by 75%, marking the first time that a therapy has been able to directly target the root cause of Huntington’s disease. This isn’t a cure; Ole Isacson, a Harvard neuroscientist, notes that patients still showed evidence of dying neurons. But slowing disease progression could lead to years of additional independence for Huntington’s patients. UniQure plans to seek approval from the US Food and Drug Administration (FDA) in 2026. But even if the therapy is approved, there will be limitations. The treatment needs to be directly and precisely inserted into the parts of the brain that are affected by Huntington’s disease – a 12 hour surgery with a probable price tag of over $1 million USD per person. Still, despite the limitations, uniQure’s clinical trial represents the first time that doctors and researchers have been able to slow Huntington’s progression. Whether it serves as a final therapy or as a launch pad for future innovations, it also represents the first hope for a group of patients who, medically, haven’t had much.
Peer Editor: Nebiyou Yonas Metaferia